
周水平
医学博士、研究员,博士毕业于北京中医药大学。现任天士力控股集团研究院执行副院长,中国药科大学、沈阳药科大学校外导师;先后主持98个新药项目研发,获得6个新药证书/生产批件、15个新药临床批件;作为发明人,先后申请发明专利205件(PCT73项),获得授权75件(PCT24项);先后主持或参与15项国家及省部级课题,发表专业论文109篇,其中SCI文章19篇、专著4部;获得国家科学技术进步二等奖1项、省部级一等奖3项、二等奖1项。
加强中医药的科技创新并实现其现代化和国际化,是我国从1996年倡导中药现代化战略行动以来一直坚持的基本国策和目标,近年来,在国家“一带一路”战略引领下,以《国家中长期科学和技术发展纲要(2006-2020年)》、《中医药事业发展“十二五”规划》、《中医药创新发展规划纲要(2016-2030年)》等一系列指导性产业政策和文件的颁布和出台为标志,为扶持我国创新中药国际化研发提供了良好的环境和氛围。
提问一:请您简要介绍一下创新中药国际化的模式和成果
面对疾病谱演变、医学模式变化以及国际新药生产力下降等现状,植物药已经成为了全球创新药物研发的重要源泉,除传统的从植物中发现单体活性成分并进行结构修饰/优化以获得新药这一途径外,定位于多种已知或未知活性成分的混合物作为新药进行创新研发,也成为国际制药行业近年来新药研发的重要途径和选择。 具有多组分多靶点治疗特点的中药,因其整体性治疗法则恰好适合了现代医学的需要。
不同于从早期新分子实体筛选/合成/修饰到临床有效性评价的西药研发模式,基于中药安全性和有效性数据,通过全面的追溯和回顾性研究而进行反向药物开发并最终实现上市,这一研发模式在美国FDA的植物药工业指南的实施精神中得到充分体现。
目前,以中医理论为基础具有人用经验的创新中药,其国际化研发可以分为两种模式:
第一种是以美国FDA为代表的渐进性、创新式的开发,基于中药的传统人用经验,创新中药可从人体临床研究重新起步,在产品开发过程中,根据研发需要进行逆向式的药理毒理和临床研究,获得批准和上市。
第二种是以欧盟传统药申请为代表的回顾性的研究,基于该品种或相似品种充足的人用经验和文献证据的评价结果,研发品种具备了产品免除大部分临床和临床研究的条件,通过完整的质量开发,就能满足此类中药产品获得批准和上市。
就欧美发达国家而言,目前正在申报美国FDA开展临床研究的中药品种已经超过了十个,复方丹参滴丸已顺利完成Ⅲ期临床,现进入COV(临床中心关闭访查)阶段;扶正化瘀片、血脂康、康莱特注射液、HMPL-004(穿心莲提取物)等已完成Ⅱ期临床。同时,中药产品地奥心血康和丹参胶囊也已在荷兰的先后成功注册,表明了欧盟药审局已开始正式接纳中药合法进入医药市场。
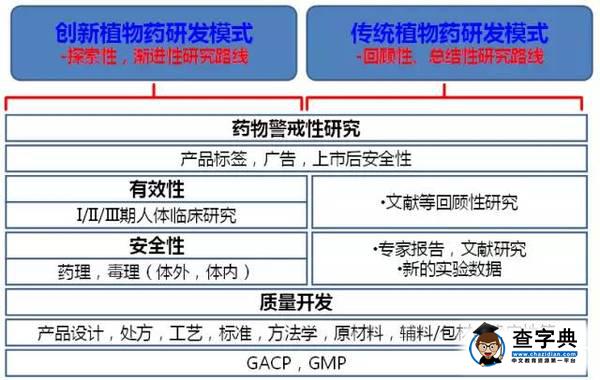
国际化研发模式比较
提问二:选择哪些中药品种进行国际化研发更有可行性呢?
以FDA申报为例,走在国际化前列的药物有这样一些共同点:
①为国内已上市多年的产品,多为中药大品种。临床应用经验丰富,安全性和有效性得到充分验证,不良反应信息较为全面。因此,得到FDA批准进行临床试验的可能性大。
②丰富的文献支持资料,包括临床前和临床研究文献,能为临床获批提供更多支持数据。如,CNKI中可搜索到接近2万条复方丹参滴丸相关文献。
③除2015年新获批Ⅱ期临床的连花清瘟胶囊为13味大复方外,其他药品组方相对简单,为1-6味药材组成,在提供符合法规要求的CMC资料上面临的挑战相对小。
④除康莱特注射液外,其余均为口服剂型。而康莱特注射液仅含单一药材提取物,组分相对简单,相较而言更容易实现良好的药品质量控制。
故而,我们的策略与建议是:
人用经验丰富的中药品种,国际申报开展Ⅱ期研究,同时根据法规要求补充I期或毒理资料;
缺乏人用经验而在临床前研究有潜力的品种,可国内国际同步申请IND,国际申报开展I期研究,国内申报走加快审批途径。
而就中药品种欧盟申报而言,符合性申请有严格的限定条件:包括剂型(只能口服、外用和喷雾剂型),适应症(OTC用途),传统年限(15/30年)等。基于法规定义, 中药产品应作为OTC产品,即患者不需医生监督可自行使用。欧盟法规要求的“15/30年应用”,在可以减免临床研究的基础上,通过多重证据链证实产品的安全性/有效性是解决问题的关键。同时,通过增补遗传毒性研究和药物上市后警戒性等额外的临床安全性数据,也是中药研发中要考虑的一个方面。
提问三:关于创新中药的国际化,可否分享一下您所在企业的成功经验?
那我从CMC研究、药理毒理研究、临床研究三个方面简要介绍一下吧:
(一)CMC研究
CMC研究是新药申请(NDA)的必要、关键环节。美国FDA、欧盟EMA均要求以正确的工艺及配套验证方法,精准执行,且验证贯穿始终,以确保生产、质量控制及技术研究体系达到法规水平。
目前中药国际化研发CMC的瓶颈在于:如何以物质基础研究和组分证明有效性、如何进行中药质量一致性评价、如何进行药品全生命周期的质量管理。
我的建议是,全产业链必须建立多组分药物质量控制标准体系:
①提倡质量源于设计:贯彻“质量源于设计(QbD)”的理念,按照ICH和FDA的指南,将药品的生产质量控制体系在研发阶段就予以设计并确认。
②基于质量风险管理:本着“基于风险”和“基于科学”两大原则,项目中的工艺变更、标准变更和设备设施的变更,都要有利于降低产品质量风险。关键质量属性(CQA)和关键工艺参数(CPP)的确定,也要基于工艺过程的风险分析。
③强化过程控制:药材、辅料、内外包装材料以及工艺过程中所用溶剂和物料,在基于风险评估基础上,采用全面质量控制,达到法规要求。
④实施全面验证:按照产品生命周期设计进行验证,从“纸工厂”到新车间建设运行,技术转移及工艺验证覆盖各个专项工作,对工作标准和逻辑性进行严格控制。
⑤遵循国际标准:按照FDA、欧盟、ICH、ISPE(国际制药工程协会)等国际化标准和指南,从临床样品生产阶段,就全面实施cGMP质量管理。
⑥从源头保障药材质量:将道地药材与数字科技相融合,从中药材交易、中药材第三方检测和产品溯源三大核心方面,按国际化要求倒逼药材质量提升。
(二)药理毒理研究
①熟悉FDA关注点,合理布局药理毒理研究:FDA对于植物药药理毒理研究采取分阶段申报原则,IND及早期临床阶段,FDA对于已上市且无不安全报道的植物产品相关非临床药理毒理研究要求不高,提供文献支持以及早期的GLP或非GLP毒理研究均可以。在进入Ⅲ期临床及NDA阶段,需要补齐相关毒理研究,包括安全药理、遗传毒性、DDI、非啮齿动物重复给药毒性、生殖毒性以及致癌性研究。
②FDA对药用辅料非临床安全性研究是非常重视,如含有新药用辅料,均需要提供新辅料支持性毒性研究。
③除规定在GLP实验室完成毒理研究实验,FDA对遗传毒、生殖毒、致癌性、辅料毒性以及毒理研究中的供试品分析和毒代研究要求较国内更为关注和严格;重视程度与化学药相同。
④参考ICH和FDA相关指南,在重复剂量毒性研究、遗传毒性研究、生殖毒性试验、致癌性试验均需要伴随TK研究。对于复杂成分中药,TK markers的选取及方法建立可采取遵循毒性成分及代谢产物、其次主要活性成分及其代谢产物、再则典型可测/易测的成分等逐层考虑原则。
⑤由于中药组分复杂性的特点,其提取物整体作为活性物质,不能简单按照化学药成分控制的质量评价方法,也不能通过BE研究去评价工艺改进等对中药产品质量的影响。FDA对中药的质量一致性评价提出更多的要求:即含有多成分及未能阐述清楚所有成分的植物药申报时均建议开发基于临床疗效机制的生物效价(Biological assay)方法用于中药产品质量一致性评价。
(三)临床研究
①适应症选择
如何选择适应症,是创新中药国际化临床研究的关键问题之一,直接决定其有效性评价。 定位于国内市场的中药品种,其适应症多为中医证侯。在国际化的临床研究中,需要选择基于西方医学理论的明确疾病或症状作为适应症。
西医依据身体系统、发病机制、病理改变、临床表现、疾病所处阶段等对于疾病有着非常详细的划分,每一种疾病又根据其临床特点进行进一步分类,有不同的治疗方案。中药在选择适应症时必须以此为依据。同时需要关注到,疾病常有发展转归,在不同阶段有不同特点及相应的治疗方案。对于相关疾病,需要明确治疗阶段。
中医的特点之一为成分复杂、多靶点起效,不同于西医成分单一、靶点明确、作用迅速的特点。在适应症的选择上,需要结合中药以及疾病特点进行充分评估。对于已有良好治疗方案的疾病、危重疾病或中医没有确切疗效的疾病等需审慎选择。此类疾病不仅有较大伦理风险,也很难在临床试验中显示出较好疗效。
②临床方案设计
中药国际临床试验的整个过程,是在了解中药基本特性的基础上,提出问题(试验目的),通过科学方法(方案设计符合GCP的试验过程)寻求答案和得到验证的逐步深入的过程。中药国际临床研发是一个循序渐进的过程,从小规模的前期试验获知药物的基本特性,到后续大规模的确证性试验,临床研发计划、目标设定和阶段性评估非常重要,有助于及时调整研发策略,减低研发风险。
临床研究质量源于顶层设计,计量统计药理学方法的应用在方案设计中发挥重要作用。给药剂量与剂量跨度,不同的给药方案,病人的群属、年龄和基础值,药效学峰谷效应因素,治疗周期、基础用药、合并用药,病人依从性等因素均需要科学综合考虑。
简而言之,科学合理的临床试验方案设计是试验成功的基础,符合GCP和ICH要求的临床试验是通向成功的阶梯,严谨的数据管理是评价药物安全性及有效性的保证。
③充分掌握药政法规
例如,SPA是FDA与企业就重大或创新性研究设立的一个特别法规程序。FDA与企业达成SPA共识后,即成为双方共同遵守的约定,在新药评审(NDA)时,将不再对达成共识的研究方案提出疑义。
另外,充分使用罕见病产品的相关的孤儿药认定、罕见病产品资助等政策;优先审评、快速通道、突破性疗法等可能的审批途径。
提问四:您对有志于中药国际化研发的企业还有哪些建议?
目前,我国冲击欧美发达国家医药市场的中药品种不断增多,但多为各品种、各企业间的单打独斗,在全行业范围内缺乏可以在一定程度上互相交流和共享的协调体系,类似行业协会的功能需要加强,为后续项目提供可供借鉴的研究经验和通路,形成足够的产业引导效应。
多数企业参与现代化国际化的意愿和热情很高,但在具体操作层面上,由于缺乏顶层设计和辅导,一些企业的现代化和国际化战略定位不清晰,风险考虑不周全,实施过程不科学,导致部分国际化品种在进展到某一阶段后,易于遭遇不必要的挫折而止步。
建议有志于中药国际化研发的企业与有丰富临床试验开展经验的国际CRO合作,寻求专家对于试验设计和临床开发方案的建议,选择合格CRO监察临床研究开展的GCP符合性、数据管理,确保合格临床研究报告的撰写及与药政部门更有效的沟通。
概而言之,创新中药国际化研发是一项宏伟的系统工程,以上建议难免挂一漏万。中药企业需考虑科研、产业、市场、资本等要素的分配,具体可参考闫凯镜博士提出的MITRO模型即“市场为导向、产业为基础、科技为引领、法规为准绳、组织为保障”,进行集成研究。
从外部环境而言,FDA、EMA也在逐步完善其对植物药的监管和开发指南,这些进步都足以看到中医药的认可程度在逐步提高;同时国家对中药国际化的鼓励政策也一直在逐步建立和实施。相信未来将有更多优秀的中药企业将勇敢地踏上国际化征程,为中药这一华夏瑰宝造福世界而努力。
作者|周水平
技术|何思航
更多精彩资讯请关注查字典资讯网,我们将持续为您更新最新资讯!
上一篇:【国医·资讯】关于遴选北京中医药大学第二... 下一篇:【多彩北化】那些与室友不得不说的秘密
 【考研快讯】这所大学多专业停招!
【考研快讯】这所大学多专业停招! 2021QS世界大学排名发布!41所中国内地高校上榜!
2021QS世界大学排名发布!41所中国内地高校上榜! 天津大学2021级研究生招生夏令营
天津大学2021级研究生招生夏令营 南京财经大学研究生院党支部与会计学院会计学师生党支部开展党建共建
南京财经大学研究生院党支部与会计学院会计学师生党支部开展党建共建 2020高考科普系列来了:国立大学与公立大学的区别
2020高考科普系列来了:国立大学与公立大学的区别 西藏民族大学2020年硕士研究生调剂公告
西藏民族大学2020年硕士研究生调剂公告 北京航空航天大学2020年优秀大学生物理夏令营通知
北京航空航天大学2020年优秀大学生物理夏令营通知 北京航空航天大学2020考研复试分数线已公布-查字典资讯网
北京航空航天大学2020考研复试分数线已公布-查字典资讯网